Über Desmoid-Tumoren
Krankheitsbild
Eine infiltrative und aggressive Erkrankung1
- Desmoid-Tumoren sind seltene Tumoren des Weichgewebes mit lokal aggressiver Proliferation, die zur Infiltration angrenzender Strukturen und zur Wucherung entlang der Faszien und Muskeln neigen1,2
- Diese zuweilen als aggressive Fibromatose oder Desmoid-Fibromatose bezeichneten3 mesenchymalen Tumoren können schwerwiegende, einschränkende und in seltenen Fällen sogar lebensbedrohliche Organschäden verursachen2
- Desmoid-Tumoren können das umliegende Gewebe infiltrieren und auf die Muskeln, Nerven und Gefäße drücken4,5
Symptomatik und Tumorlokalisation
Desmoid-Tumoren können zu schweren gesundheitlichen Beeinträchtigungen führen2
- Die Symptomatik von Desmoid-Tumoren variiert je nach Tumorlokalisation5,6
- Eine prospektive Kohortenstudie ergab, dass Tumoren in der Brustwand, in den oberen Extremitäten und im Kopf- und Nackenbereich mit schlechteren postoperativen Prognosen assoziiert waren.7
| TUMORLOKALISATION | GESCHÄTZTE HÄUFIGKEIT5 | HÄUFIGE SYMPTOME UND KOMPLIKATIONEN |
|---|---|---|
| Intraabdominell (einschließlich Mesenterium) | 20% | Kompression kann Schmerzen, Kachexie, Unwohlsein, Blähbauch oder Verstopfung des Darms oder der Harnleiter verursachen5,8 |
| Bauchwand | 16% | Große Tumoren können Gewebedehnung, Schwäche und Darm- oder Blasenverschiebung verursachen9 |
| Untere Extremitäten | 16% | Eingeschränkte Beweglichkeit, Schmerzen, Muskelsteifheit oder Fehlstellungen5,10 |
| Brustwand | 15% | Atemnot,5 Schluckstörung,5 Pleurainvasion,8 Wirbelsäulen-11 oder Rippenbeteiligung,12 Knochenerosion12 und Schmerzen12 |
| Obere Extremitäten | 14% | Eingeschränkte Beweglichkeit, Muskel- und Bänderbeteiligung, Schwäche der Extremitäten, Fehlstellungen oder Schmerzen5,13 |
| Kopf und Hals | 8% | Schmerzen, neurologische Defizite, Nähe zu vitalen Strukturen, einschließlich Mortalitätsrisiko durch Gefäß- oder Atemwegsverengung14 |
| Sonstige | 11% | Lokalisationsbedingte Symptome und Komplikationen5,6 |
Die Lokalisation der Desmoid-Tumoren kann erhebliche Auswirkungen auf die Lebensqualität haben.15 Ein Desmoid-Tumor mit „rankenartigem“ Wachstum1, bei dem die Nerven umschlossen werden, kann bspw. mit einschränkenden neuropathischen Schmerzen verbunden sein.5,16
Krankheitsverlauf
Variabel und unvorhersehbar1
- Desmoid-Tumoren zeichnen sich durch einen variablen und unvorhersehbaren Krankheitsverlauf aus, der von der Tumorlokalisation und den damit verbundenen gesundheitlichen Beeinträchtigungen abhängt1,5
- Etwa 50 % der Desmoid-Tumoren zeigen eine aggressive Biologie mit weiterem Wachstum oder Symptombildung. Die Tumorprogressionen traten größtenteils (89 %) innerhalb der ersten zwei Jahre der Beobachtung auf.4
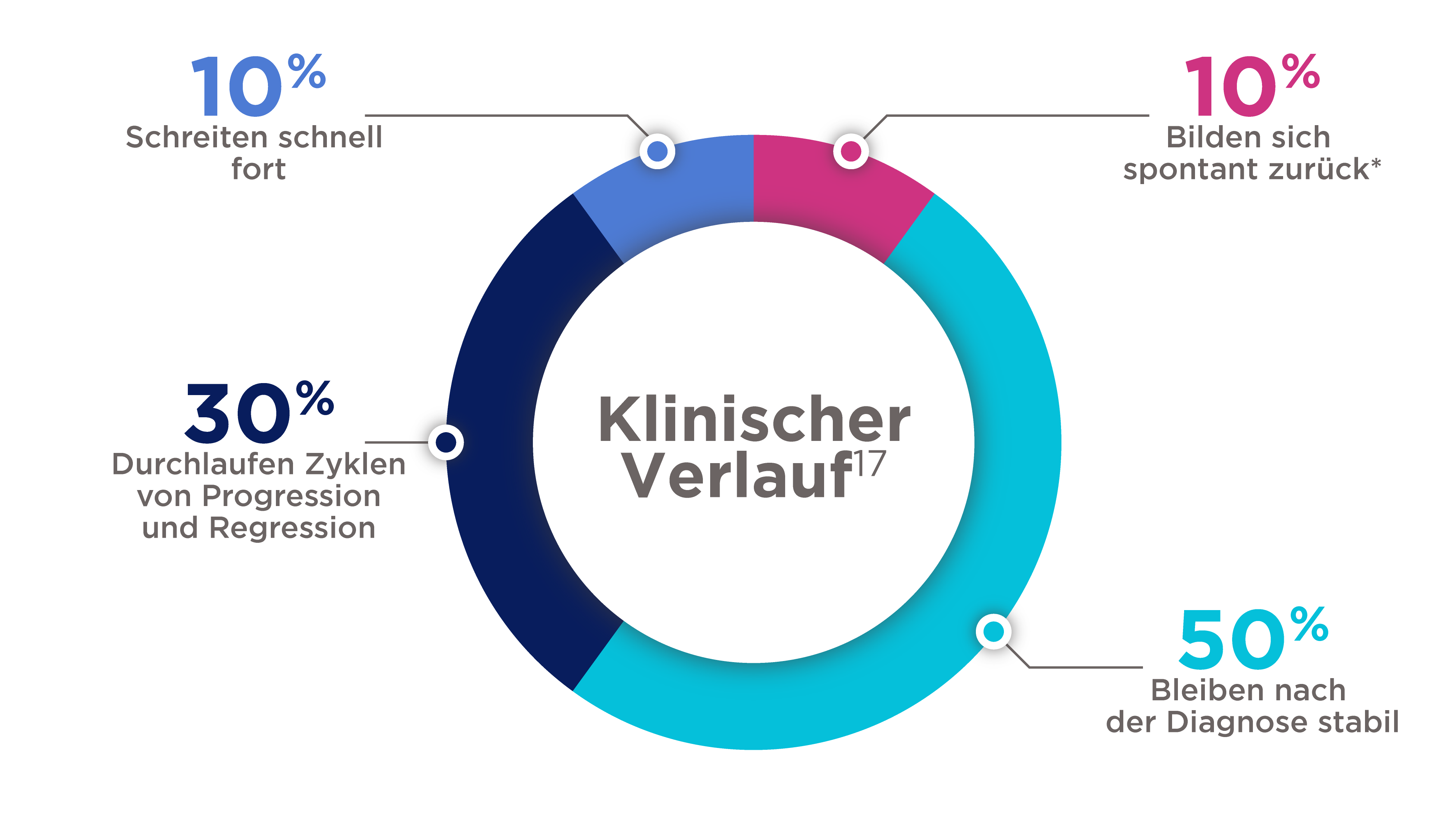
Klinischer Verlauf von Desmoid-Tumoren
*10–28% der Desmoid-Tumoren bilden sich ohne Behandlung spontan zurück17
Inzidenz und Risikofaktoren
Epidemiologie
Desmoid-Tumoren haben eine Inzidenz von etwa 5 Fällen pro 1 Millionen Einwohner pro Jahr.18
Die Orphanet-Berichtsreihe gibt eine durchschnittliche Inzidenz von 3 Fällen von Desmoid-Tumoren pro 1 Million Einwohner pro Jahr in Europa an.17
Epidemiologische Studien offenbaren, dass die meisten Desmoid-Tumor-Fälle in der Altersgruppe der 20- bis 44-jährigen auftreten.17
- Desmoid-Tumoren treten bei Kindern und älteren Erwachsenen seltener auf.19
- Die geschätzte Inzidenz ist in den vergangenen Jahren gestiegen, was auf bessere Diagnoseverfahren zurückzuführen sein könnte.17
Weibliches Geschlecht
Frauen sind 2- bis 3-mal häufiger betroffen als Männer.20
- Eine Schwangerschaft kann Desmoid-Tumoren begünstigen4
Vorangegangene Traumata
Etwa 25 % der Patientinnen und Patienten mit Desmoid-Tumoren weisen eine Anamnese mit vorangegangenen Traumata auf.5
- Verletzungen oder chirurgische Behandlungen können das Risiko erhöhen.21
APC (Adenomatöse Polyposis Coli-Gen)-Mutationen
Phänotypische Charakteristika korrelieren mit der Position der APC-Mutation im Verhältnis zu bestimmten Codons.22
- Patientinnen und Patienten mit familiärer adenomatöser Polyposis (FAP) haben ein 1000-faches höheres Risiko Desmoid-Tumoren zu entwickeln, als die Allgemeinbevölkerung.20
ICD-10-CM-Codes für Desmoid-Tumoren
Ätiologie und Pathogenese von Desmoid-Tumoren
Das Wissen um die molekulare Pathogenese ist in Anbetracht des äußerst variablen klinischen Verlaufs dieser Erkrankung von erheblichem Interesse19
Die Ätiologie von Desmoid-Tumoren ist unbekannt. Die Identifizierung klonaler Chromosomenveränderungen in einer erheblichen Zahl von Fällen untermauert jedoch die neoplastische Natur dieser Tumoren.19
- Man geht davon aus, dass die Pathogenese von Desmoid-Tumoren mit Traumata im Zusammenhang steht4
- Chirurgische Behandlungen können das Tumorwachstum begünstigen4
- Wachstumsfaktoren, die während der ersten Wundheilungsphase nach der chirurgischen Behandlung freigesetzt werden, können Signale übertragen, die die Aktivierung von β-Catenin fördern.4
- Eine Schwangerschaft kann Desmoid-Tumoren nicht nur sekundär durch hormonelle Einflüsse begünstigen, sondern auch durch die systemische Freisetzung von Wachstumsfaktoren.4
- Es sind zwei Signalwege beteiligt: 1) der Wnt/ß-Catenin/APC-Signalweg, bei dem CTNNB1- und APC-Mutationen zu einer ß-Catenin-Akkumulation führen, und 2) der Notch-Signalweg, bei dem zwei Spaltungen an Notch-Rezeptoren auftreten.17
- Die potenzielle Wechselwirkung zwischen den Notch- und Wnt-Signalwegen sowie die Aktivierung des Notch-Signalwegs infolge der Fehlregulation des Wnt-Signalwegs scheinen an der Pathogenese von Desmoid-Tumoren beteiligt zu sein.17
Nukleäre Akkumulation von β-Catenin
- An der Pathogenese von Desmoid-Tumoren ist eine Fehlregulation des Wnt/β-Catenin-Signalwegs beteiligt2,23
- Die nukleäre Akkumulation von β-Catenin und die nachgeschaltete Gentranskription ist ein wesentlicher Treiber für die Proliferation von Desmoid-Tumorzellen2,24
- Die nukleäre Akkumulation von β-Catenin kann initiiert werden durch2,24
- Aktivierende Mutationen im β-Catenin-Gen CTNNB1
- Inaktivierende Mutationen im negativen Regulator APC, häufig im Rahmen einer familiären adenomatösen Polyposis (FAP)
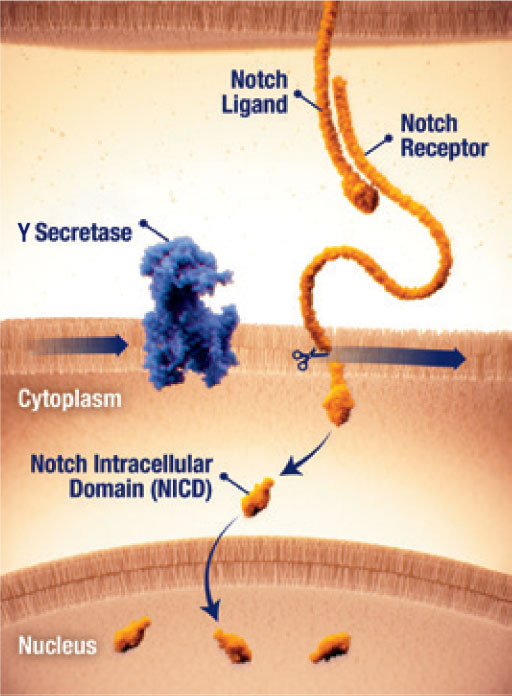
Notch-Aktivierung durch γ-Sekretase
- Der Notch-Signalweg kann bei Desmoid-Tumoren aktiv sein.25
- Eine Fehlregulation des Notch-Signalwegs kann zur Aktivierung von Signalwegen führen, die zum Tumorwachstum beitragen.25
- Für die Signalübertragung des Notch-Rezeptors ist eine proteolytische Aktivierung durch das Enzγm γ-Sekretase (Gamma-Sekretase) erforderlich.25
- Durch die Spaltung durch die Gamma-Sekretase wird die intrazelluläre Notch-Domäne (NICD) freigelegt, welche zum Zellkern transloziert und die Gentranskription aktiviert.25
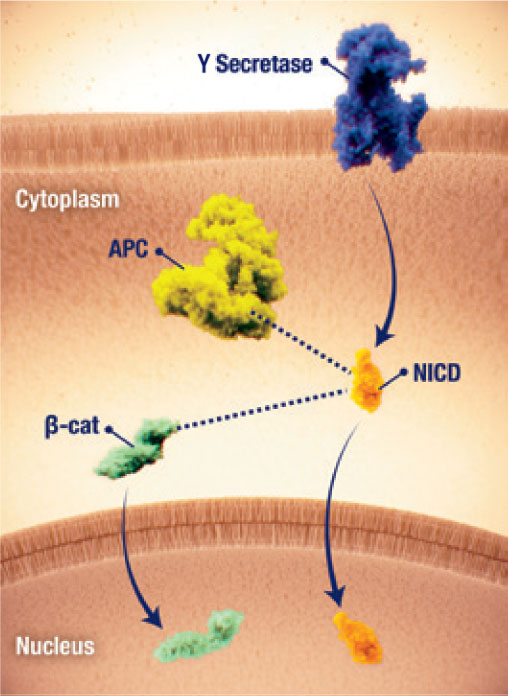
Wechselwirkung von Signalwegen
- Eine Wechselwirkung zwischen den Wnt/β-Catenin- und Notch-Signalwegen könnte zur weiteren Desmoid-Tumor-Pathogenese beitragen.17
- An der Pathogenese von Desmoid-Tumoren ist eine Fehlregulierung des Wnt/β-Catenin-Signalwegs beteiligt.2,24
- Die nukleäre Akkumulation von β-Catenin und die nachgeschaltete Gentranskription ist ein wesentlicher Treiber für die Proliferation von Desmoid-Tumorzellen.2,25
- Die nukleäre Akkumulation von β-Catenin kann initiiert werden durch.2,25
- Aktivierende Mutationen im β-Catenin-Gen CTNNB1
- Inaktivierende Mutationen im negativen Regulator APC, häufig im Rahmen einer familiären adenomatösen Polyposis (FAP)
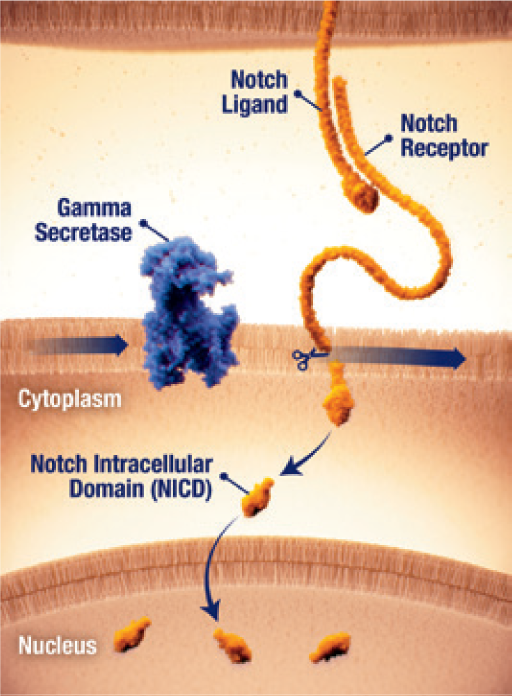
- Der Notch-Signalweg kann bei Desmoid-Tumoren aktiv sein.26
- Ein fehlreguliertes Notch-Protein kann Signalwege aktivieren, die zum Tumorwachstum beitragen.26
- Für den Signalweg des Notch-Rezeptors ist eine proteolytische Aktivierung durch das Enzγm Gamma-Sekretase erforderlich.26
- Durch die Spaltung der γ-Sekretase wird die intrazelluläre Notch-Domäne (NICD) freigelegt, welche zum Zellkern transloziert und die Gentranskription aktiviert.26

- Eine Wechselwirkung zwischen den Wnt/β-Catenin- und Notch-Signalwegen kann zur weiteren Desmoid-Tumor-Pathogenese beitragen.18
Rezidivrisiko
- Die Rezidivraten können von der Tumorlokalisation beeinflusst sein:17
- Tumoren an den Extremitäten werden als lokal aggressiv eingestuft und haben Rezidivraten von 24 % bis 77 %.
- Die lokalen Rezidivraten von intraabdominellen Tumoren bei Patientinnen und Patienten mit FAP (Familiärer Adenomatöser Polyposis) sind höher als die für extraabdominelle Tumoren und werden mit 57 % bis 86 % angegeben.
Nach einem chirurgischen Eingriff besteht ein hohes lokales Rezidivrisiko17
- Die Rezidivraten können durch Traumata, wie z. B. durch chirurgische Eingriffe, verschlechtert werden und liegen nach 5 Jahren zwischen etwa 25 % und 60 %.17
- Die Invasion wichtiger Gefäße und Nerven und die Resektionsränder sind maßgebliche Faktoren für die hohe postoperative Rezidivrate26
- Unglücklicherweise sind die Rezidivraten selbst bei tumorfreien Resektionsrändern hoch27
Wachstumsfaktoren, die während der ersten Wundheilungsphase nach dem chirurgischen Eingriff freigesetzt werden, könnten die β-Catenin-Aktivierung fördern.4
- Die Tatsache, dass manche Tumoren nach einem chirurgischen Eingriff erneut auftraten, anschließend jedoch ohne Behandlung stabil blieben, legt nahe, dass nach dem chirurgischen Eingriff freigesetzte Wachstumsfaktoren das erneute Auftreten von Tumoren gefördert haben könnten, die ansonsten inaktiv geblieben wären.4
- Die spezifische CTNNB1-Mutation S45F scheint mit einer schlechteren rezidivfreien Überlebensrate nach der chirurgische Behandlung assoziiert zu sein.28
- Die rezidivfreie Überlebensrate nach 5 Jahren war bei Patientinnen und Patienten mit S45F-mutierten Desmoid-Tumoren signifikant schlechter (P<0,0001) im Vergleich zu Patientinnen und Patienten mit T41A-mutierten oder nicht mutierten Tumoren.29
| MUTATION | GESCHÄTZTES REZIDIVFREIES ÜBERLEBEN (95 % CI) |
|---|---|
| 45F | 23% (10%-40%) |
| 41A | 57% (43%-69%) |
| Nicht-mutiertes CTNNB1 | 65% (38%-83%) |
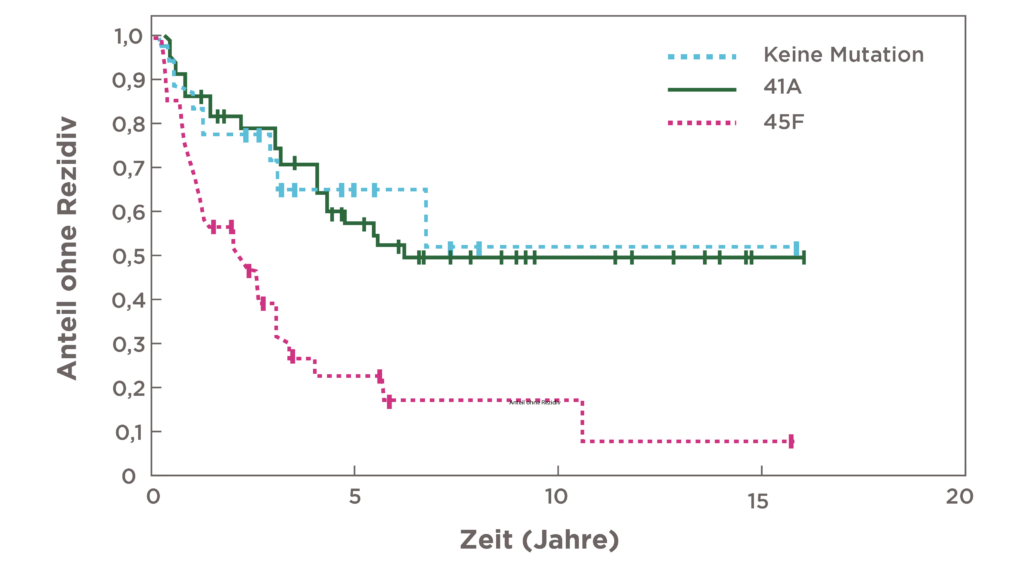
Patientinnen und Patienten mit CTNNB1-Mutationen bei Codon 45 (45F) haben ein dreifach höheres Rezidivrisiko nach der Operation29
Im Rahmen einer Metaanalyse wurde die Tumorgröße als wichtiger Mediator für ein erhöhtes Rezidivrisiko bei Patienten mit der CTNNB1-Mutation S45F genannt.30
Behandlungsgrundsätze
Eine sorgfältige Auswahl der Behandlungsstrategie für die jeweiligen Patientinnen und Patienten mit Desmoid-Tumor ist unverzichtbar für eine optimale Tumorkontrolle und Verbesserung der Lebensqualität18
Im Laufe des vergangen Jahrzehntes wurden erhebliche Anstrengungen für eine standardisierte Behandlung dieser Erkrankung unternommen.18
Der derzeit unerfüllte Bedarf in Bezug auf Desmoid-Tumoren umfasst eine frühzeitige und genaue Diagnose sowie zugelassene Behandlungen für Patientinnen und Patienten mit Desmoid-Tumoren.17
Im Rahmen einer am Memorial Sloan Kettering Cancer Center durchgeführten Studie gingen 58 % der Patientinnen und Patienten mit Desmoid-Tumor von der Active Surveillance in eine Erstbehandlung über, ohne dass eine radiologische Tumorprogression auftrat.31†
Die Behandlungsziele sollten sich nicht nur auf klinische Marker wie das progressionsfreie Überleben konzentrieren, sondern auch patientinnen- und patientenrelevante Endpunkte berücksichtigen, wie zum Beispiel eine Reduktion der Desmoid-Tumor-spezifischen Symptomlast (z. B. Schmerz) und deren Auswirkungen auf das Leben der Patientinnen und Patienten, die Verbesserung der Alltagsfunktionen und die allgemeine Lebensqualität.17
Früherkennung der Progression
Die Erkennung der Desmoid-Tumorprogression kann zur rechtzeitigen Einleitung einer angemessenen Behandlung führen.
- Um die Patientinnen- und Patientenoutcomes zu verbessern, sollten medizinische Fachkräfte eine Progression möglichst früh anhand mindestens eines der folgenden Kriterien beurteilen:
- Schmerz kann ein prognostischer Indikator für ein Fortschreiten der Erkrankung sein34 und mit einem schlechteren Krankheitsverlauf einhergehen35
- Bei größeren Tumoren kann ein größeres Risiko für ein Fortschreiten der Erkrankung bestehen3,33
†Patientinnen und Patienten mit erstmals oder erneut auftretenden Desmoid-Tumoren (n=160) wurden retrospektiv anhand einer institutionellen Datenbank ermittelt. Unter den erstmalig beobachteten Patientinnen und Patienten, für die serielle MRT-Untersuchungen zur Verfügung standen, waren 14 von 24 (58 %) Patientinnen und Patienten mit einer aktiven Behandlung nicht von einem Tumorwachstum gemäß Definition nach den RECIST-Kriterien betroffen.31 Modifiziert nach Text, Referenz 31.
Hören Sie sich die medizinischen Meinungen von Onkologen zu Desmoid-Tumoren an
Desmoid-Tumoren verstehen
Schauen Sie sich das Video von Dr. Riedel, medizinischer Onkologe vom Duke Health and Duke Cancer Institute, mit einer Übersicht über Desmoid-Tumoren an, inklusive der Pathophysiologie, dem klinischen Bild und den Auswirkungen für die Patientinnen und Patienten.
APC: adenomatöses Polyposis Coli-Gen; CT: Computertomografie; CTNNB1: Catenin-β1-Gen; FAP: familiäre adenomatöse Polyposis; MRT: Magnetresonanztomographie; NICD: intrazelluläre Notch-Domäne; RECIST: Response Evaluation Criteria In Solid Tumors; S45F: Mutationsart; T41A: Mutationsart; Wnt: Wingless-assoziierte Integrationsstelle.
Referenzen
- Kasper B et al. Desmoid Working Group. An update on the management of sporadic desmoid-type fibromatosis: a European Consensus Initiative between Sarcoma Patients EuroNet (SPAEN) and European Organization for Research and Treatment of Cancer (EORTC)/Soft Tissue and Bone Sarcoma Group (STBSG). Ann Oncol. 2017;28(10):2399-2408.
- Penel N et al. Adult desmoid tumors: biology, management and ongoing trials. Curr Opin Oncol. 2017;29(4):268-274.
- Gronchi A et al. Desmoid Tumor Working Group. The management of desmoid tumours: a joint global consensus-based guideline approach for adult and paediatric patients. Eur J Cancer. 2020;127:96-107.
- Bonvalot S et al. The treatment of desmoid tumors: a stepwise clinical approach. Ann Oncol. 2012;23(erg. 10):x158-x166.
- Constantinidou A et al. Clinical presentation of desmoid tumors. In: Litchman C, ed. Desmoid Tumors. Springer; 2012: Kap. 2. Abgerufen am 9. April 2024. https://www.researchgate.net/publication/226455135.
- Joglekar SB et al. Current perspectives on desmoid tumors: the mayo clinic approach. Cancers (Basel). 2011;3(3):3143-3155.
- Penel N et al. Surgical versus non-surgical approach in primary desmoid-type fibromatosis patients: a nationwide prospective cohort from the French Sarcoma Group. Eur J Cancer. 2017;83:125-131.
- Shinagare AB et al. A to Z of desmoid tumors. AJR Am J Roentgenol. 2011;197(6):W1008-W1014.
- Koshariya M et al. Giant desmoid tumor of the anterior abdominal wall in a young female: a case report. Case Rep Surg. 2013;2013:780862.
- McDonald ES et al. Best cases from the AFIP: extraabdominal desmoid-type fibromatosis. Radiographics. 2008;28(3):901-906.
- Abrão FC et al. Desmoid tumors of the chest wall: surgical challenges and possible risk factors. Clinics (Sao Paulo). 2011;66(4):705-708.
- Xie Y et al. Recurrent desmoid tumor of the mediastinum: a case report. Oncol Lett. 2014;8(5):2276-2278.
- Scaramussa FS et al. Desmoid tumor in hand: a case report. SM J Orthop. 2016;2(3):1036.
- Baranov E et al. Soft tissue special issue: fibroblastic and myofibroblastic neoplasms of the head and neck. Head Neck Pathol. 2020;14(1):43-58.
- Ingley KM et al. High prevalence of persistent emotional distress in desmoid tumor. Psychooncology. 2020;29(2):311-320.
- Gounder MM et al. Prospective development of a patient-reported outcomes instrument for desmoid tumors or aggressive fibromatosis. Cancer. 2020;126(3):531-539.
- Bektas M et al. Desmoid tumors: a comprehensive review. Adv Ther. 2023;40(9):3697-3722.
- Kasper B et al. Desmoid Tumor Working Group. Current management of desmoid tumors: a review. JAMA Oncol. 2024;10(8):1121-1128.
- Ravi V et al. Desmoid tumors: epidemiology, molecular pathogenesis, clinical presentation, diagnosis, and local therapy Jun 2024 UptoDate.
- Skubitz KM. Biology and treatment of aggressive fibromatosis or desmoid tumor. Mayo Clin Proc. 2017;92(6):947-964.
- Lopez R et al. Problems in diagnosis and management of desmoid tumors. Am J Surg. 1990;159(5):450-453.
- Kasper B et al. Desmoid tumors: clinical features and treatment options for advanced disease. Oncologist. 2011;16(5):682-93.
- Crago AM et al. Near universal detection of alterations in CTNNB1 and Wnt pathway regulators in desmoid-type fi bromatosis by whole-exome sequencing and genomic analysis. Genes Chromosomes Cancer. 2015;54(10):606-615.
- Gronchi A et al. Desmoid Tumor Working Group. The management of desmoid tumours: a joint global consensus-based guideline approach for adult and paediatric patients [ergänzender Anhang]. Eur J Cancer. 2020;127:96-107.
- Shang H et al. Targeting the Notch pathway: a potential therapeutic approach for desmoid tumors. Cancer. 2015;121(22):4088-4096.
- Wang YF et al. Postoperative recurrence of desmoid tumors: clinical and pathological perspectives. World J Surg Oncol. 2015;13:26.
- Easter DW et al. Recent trends in the management of desmoid tumors. Summary of 19 cases and review of the literature. Ann Surg. 1989;210(6):765-769.
- Napolitano A et al. Recent advances in desmoid tumor therapy. Cancers (Basel). 2020;12(8):2135.
- Lazar AJ et al. Specific mutations in the beta-catenin gene (CTNNB1) correlate with local recurrence in sporadic desmoid tumors. Am J Pathol. 2008;173(5):1518-27.
- Timbergen MJM et al. The prognostic role of β-Catenin mutations in desmoid-type fibromatosis undergoing resection only: a meta-analysis of individual patient data. Ann Surg. 2021;273(6):1094-1101.
- Cassidy MR et al. Association of MRI T2 signal intensity with desmoid tumor progression during active observation: a retrospective cohort study. Ann Surg. 2020;271(4):748-755.
- Referenziert mit der Genehmigung von NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) for Soft Tissue Sarcoma V.1.2024. © National Comprehensive Cancer Network, Inc. 2024. Alle Rechte vorbehalten. Abgerufen am 29. April 2024. Die aktuelle und vollständige Fassung der Leitlinie ist online unter NCCN.org zu finden. NCCN übernimmt keine Garantien jeglicher Art in Bezug auf die jeweiligen Inhalte, die Verwendung oder die Anwendung und lehnt jegliche Verantwortung für die jeweilige Anwendung oder Verwendung ab.
- Kasper B et al. Desmoid Tumor Working Group. Current management of desmoid tumors: a review. [ergänzender Anhang] JAMA Oncol. 2024;10(8):1121-1128.
- Cuomo P et al. Extra-abdominal desmoid tumor fibromatosis: a multicenter EMSOS study. BMC Cancer. 2021;21(1):437.
- Penel N et al. Pain in desmoid-type fibromatosis: prevalence, determinants and prognosis value. Int J Cancer. 2023;153(2):407-416.